
Our bodies consist of over 200 different types of cell. Each cell type looks and functions differently from one another, yet they all originated from the same fertilised egg and therefore share the same genetic code. Clearly, the remarkable diversity of cells must be generated independently from their genetic code. These observations can now be explained by an exciting new branch of biology – epigenetics – the study of how the cellular material that sits outside the genome is able to influence gene activity.
Underpinning epigenetics is an array of over 100 possible chemical modifications to DNA and associated proteins. Each modification can act as a signal, either individually or synergistically, to switch on or off gene activity. In this way, epigenetic modifications provide an additional layer of regulatory information over the genome, leading to many possible interpretations of the same genetic code. This can explain a conundrum that has baffled scientists – how genetically identical twins, for example, often look dissimilar and have varying disease susceptibility despite sharing the same genetic code.
Epigenetic modifications
The best characterised epigenetic modification is DNA methylation, which occurs predominantly on the carbon 5 position of cytosine (5-methylcytosine or 5mC) and accounts for ~2% of all DNA base pairs. The presence of 5mC is now recognised as the main epigenetic process through which gene activity is silenced in our genomes.
For many years, DNA methylation was assumed to be a highly stable and long-lived epigenetic modification. At an early stage of development – approximately one week after fertilisation in mice – genomes start to become modified via DNA methylation, which leads to the silencing of genes that are not required for development. As cells grow and divide, methylated DNA is copied to daughter cells, ensuring that epigenetic information is passed on to a new generation of cells. This is important because, without the inheritance of epigenetic information, it is likely that individual genes would become active in newly formed cells and this could lead to deleterious effects.
An important example of this effect was identified in the 1990s by Evani Viegas-Pequignot at the National Institute of Health in Paris, France, and Stanley Gartler at the University of Washington, Seattle, US. They found that individuals with mutations in a key enzyme that is required to methylate DNA showed reduced DNA methylation levels in several genes and an inability to silence those genes. These individuals suffered from a deficient immune system and facial anomalies, demonstrating the importance of maintaining appropriate DNA methylation during development.
In the majority of individuals, DNA methylation is a stable epigenetic modification. But, every now and again, mistakes are made during the copying of epigenetic information during cell division. Many of these isolated incidents can be tolerated by cells and seem to have little consequence on gene activity. Genomes go to great lengths to ensure that epigenetic errors are not passed to future generations and therefore nearly all methylated DNA is erased from the genome during the formation of sperm and egg cells, and also again within hours after fertilisation. This ensures a clean slate for the next generation but posed a dilemma for scientists trying to understand epigenetics – to remove the methyl group from cytosine would require the breaking of a strong carbon–carbon bond.
After many years of searching, scientists have recently discovered two new multistep biochemical processes that have the potential to remove 5mC from our genomes. In one, Anjana Rao and colleagues at Harvard Medical School, US,1 demonstrated that the enzyme TET1 could oxidise 5mC to 5-hydroxymethylcytosine (5hmC). As 5hmC is functionally different to 5mC and cannot recruit repressive binding proteins to silence gene activity, the current hypothesis is that 5hmC may be an intermediate step in the active removal of 5mC. In another, several research groups have reported biochemical evidence showing that specific enzymes can deaminate 5mC to thymine, thus providing a second potential pathway for active DNA demethylation. At present, it remains unclear the extent to which these two multistep processes are occurring in our cells and whether there is any overlap between the two pathways.
These recent discoveries have generated a great deal of excitement, not simply because they finally provide an explanation for the removal of DNA methylation, but, more broadly, they provide fundamental understanding about how our genomes are regulated. They may eventually lead to improvements in our ability to manipulate gene activity in cells and potentially to reverse disease by jump-starting a gene that has been silenced through DNA methylation.
Another major process through which epigenetic information can influence gene activity in cells is by modifying the proteins that associate closely with DNA. DNA is wrapped around histones, a family of proteins that can influence how the genome is interpreted. Histones are constantly undergoing biochemical reactions – acetylation, methylation, phosphorylation etc – within the nucleus of cells. Each modification can contribute to different functional outcomes, for instance, in altering gene activity, or marking the underlying DNA for repair. This complexity led David Allis at The Rockefeller University, US, to hypothesise that combinations of histone modifications – the ‘histone code’ – regulate our genome in an ordered and predictable manner.2 Amazingly, this implies that if we could decipher the histone code, then we could use that information to uncover new drug targets and selectively control gene activity.
Numerous scientists in Europe and the US have contributed to unravelling the complexity of this code. From their work, we now know, for example, that acetylation of histones occurs typically on lysine residues within the histone tails through acetyltransferase enzymes; an event associated with activation of gene activity. Acetylation exerts this effect by altering the overall charge on DNA and thus its conformation and accessibility to regulatory proteins, and also by generating a specific docking site for other proteins. Removal of acetylation is carried out by histone deacetylases and is associated with silencing of gene activity.
Methylation of histone proteins is another well-characterised epigenetic modification and is associated with gene activation or repression (Figure 1).
A major step forward in our understanding of the biochemical mechanisms that control histone methylation was achieved in 2004 by Yang Shi at Harvard Medical School, US, when his group identified the first enzyme capable of histone demethylation.3 This was important because, until then, methylation had long been considered a permanent modification that could not be removed from histone proteins.
Two years later, further evidence for this mode of regulation was provided by Yi Zhang, currently at Harvard Medical School, US, who discovered a second class of histone demethylase enzymes.4 The two classes of enzymes remove methylation from histone proteins using different biochemical pathways. In one, an amine oxidation reaction is the critical step, and in the other, the methyl group is hydroxylated. Together, these discoveries tell us that histone methylation is dynamically regulated by both histone methylases and demethylases and this is important for deciphering how the histone code controls our genome.
The overall epigenetic output of an individual gene, therefore, is determined by a combination of chemical modifications to DNA and to histone proteins and must be ‘read’ as a complete set of instructions. This level of understanding is critical to understand the links between epigenetic information and genome regulation, and potentially for scientists to modulate epigenetic processes to control gene activity.
Epigenetics and disease
Epigenetic modifications to DNA and histone proteins have important functions in regulating gene activity within our cells. It is not surprising, therefore, that numerous diseases are associated with loss of epigenetic control. In particular, abnormal patterns of DNA methylation are associated with several types of disease, including most human cancers. It is thought that disease may develop when errors in DNA methylation silence the activity of particular genes. Such genes are typically referred to as ‘tumour-suppressors’ because their function in healthy cells is to prevent the onset of cancer. It is not clear what triggers the silencing of tumour-suppressor genes, though recent evidence suggests that lifestyle choices such as smoking and poor diet can change DNA methylation patterns, leading to silencing of gene activity.
It is not all bad news, however. Scientists are now learning to manipulate epigenetic modifications, by developing drugs that treat disease by reactivating silenced tumour-suppressor genes. This is cause for optimism because, unlike genetic mutations that cannot be corrected, epigenetic mutations can potentially be reversed leading to a genome that is indistinguishable from the pre-disease state.
Two drugs that target DNA methylation are currently used to treat leukaemia-type diseases. Both are cytosine analogues: 5-azacytidine (Vidaza) and 5-aza-2’-deoxycytidine (Dacogen), and their therapeutic basis is thought to be due to the removal of methylated DNA from tumour-suppressor genes, leading to reactivation of their gene activity. Encouragingly, patients treated with 5-azacytidine live for roughly two years on average, compared with 15 months for those on conventional therapy.
Small molecules that inhibit histone deacetylase enzymes are also in clinical use to treat leukaemia-type diseases, including vorinostat (Zolinza) and romidepsin (Istodax). Several alternative histone deacetylase inhibitors are in clinical trails for the treatment of other cancers, such as ovarian (belinostat) and lung (entinostat). There is also evidence that epigenetic therapies are more likely to be effective when used in combination. For example, Gregory Otterson and colleagues at Ohio State University, US, demonstrated that the ability of the histone deacetylase inhibitor trichostatin A to kill lung cancer cells was greatly enhanced in the presence of 5-aza-2’-deoxycytidine, suggesting that DNA methylation may play an important role in the efficacy of histone deacetylase inhibitors.
Current limitations of epigenetic therapy include the lack of specificity, so that inhibition of DNA methylation or histone acetylation pathways are likely to alter the entire genome in all cells and therefore lead to side effects. In addition, given the probable lability of epigenetic modifications, it is possible that epigenetic states once corrected may revert back to the diseased state.
Despite these concerns, there is optimism that many of these obstacles can be overcome with improved understanding of epigenetic processes. The great hope for ongoing epigenetic research is that with the flick of a biochemical switch, we could correct genes that play a role in many other diseases.
Future developments
An exciting branch of epigenetic research is focused on understanding and applying epigenetic information to stem cell biology and regenerative medicine.
Stem cells are unspecialised, self-renewing cells that can be grown in the laboratory and when given the appropriate cue – by changing their signalling environment with growth factors and cytokines, for example – specialise into any cell type of the adult body. This remarkable property has generated a great deal of interest in stem cells for the study of human development, their potential use in patient-specific cell-replacement therapies, and other applications, including disease modelling, drug screening and toxicology assays.
Our research group uses stem cells to study epigenetic modifications because during stem cell specialisation the pattern of epigenetic modifications are rapidly altered and this is accompanied by changes in gene activity and cell state. As far as we can tell, these changes in epigenetic modifications use the same pathways that occur during the formation of tissues and organs, therefore results should be germane to processes during development. Furthermore, because stem cells are grown in a Petri dish, they are easier to observe and manipulate than tissues in developing embryos.
In particular, scientists are interested in understanding more about how histone methylation is involved in controlling gene activity during stem cell specialisation. We do this by monitoring which genes are modified by histone methylation and then look at what happens to the activity of those genes when we prevent them from acquiring histone methylation. As many of these genes are important for stem cell specialisation, we predict that this alteration will lead to defects in the ability of stem cells to specialise properly. In the long-term, we hope to use this epigenetic information to control the specialisation of stem cells into clinically useful cell types, such as heart and liver cells.
Remarkably, the reverse of stem cell specialisation is also possible, so that an adult cell can be reprogrammed into a stem cell and this process is also accompanied by a re-setting of epigenetic information. However, despite rapid progress in this field, cell reprogramming remains fairly inefficient. The removal and re-establishment of epigenetic modifications is a limiting factor probably because most epigenetic modifications are stable and resist being altered. However, small molecules that interfere with epigenetic function, such as the histone deacetylase inhibitor valproic acid, can improve reprogramming efficiency by over 100-fold.
Although we do not know exactly why the inhibitors have this effect, it is likely that they lead to increased activity of several genes, some of which have a positive role in promoting reprogramming. Like epigenetic therapies, however, current inhibitors can have undesirable side effects and therefore more focused strategies are needed to drive this research area forward.
Overall, while genetic information remains fundamental in regulating all cellular functions, it is now clear that we also need to be able to decode a new language, the array of chemical modifications that combine to form epigenetic information. Improved understanding of epigenetic information will lead to better control over genome function, better early predictors of disease and better treatments for a wide range of diseases.
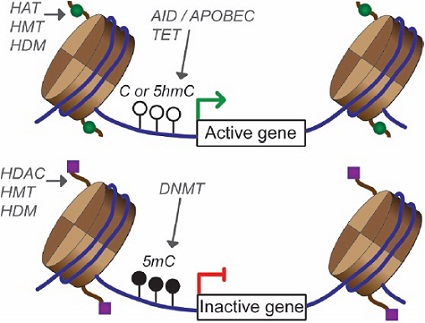
Figure 1: Chemical modifications to histones generates epignetic signals that provide an additional layer of regulation over the genome.
Peter Rugg-Gunn is group leader of the epigenetics programme at the Babraham Institute, Cambridge, UK.



