
Recent inventions have expanded the range of biomolecular targets that can be imaged non-destructively with visible light. Michael Gross reports
The nanoscale world within a living cell has long remained hidden from view, as the wavelength of the light we use for vision limits what we can see. According to a physical law discovered by German physicist and mathematician Ernst Abbe (1840-1905), microscopy can resolve two points only if they are at least half a wavelength apart, so everything below 200nm falls beyond this socalled diffraction limit.
Or so we thought for over a century. In recent years, physicists have developed a growing toolkit of instruments that use visible light and still achieve resolution better than 100nm, allowing researchers to peer deep into the nanoworld, which is important both for our understanding of the living cell and for the advancement of nanotechnology. How is this possible?
Spot on

‘In STED, a normal excitation spot is overlaid with the quenching doughnut shaped spot, which allows the emission of fluorescence photons only from the centre of the excitation spot,’ Rizzoli explains. The second laser de-excites molecules in the outer parts of the excitation spot, leaving only molecules in the middle with the energy needed to fluoresce. The de-excitation is based on stimulated emission, a phenomenon first described by Einstein in 1917, which is also fundamental to the function of the laser itself. While this process also produces light, Rizzoli says ‘this is a longer wavelength than that of the normally emitted photons, and can be simply taken away by conventional fluorescence filters.’ By depleting fluorescence in the outer ring of the excitation spot, one can reduce the effective spot size to as little as 25nm.
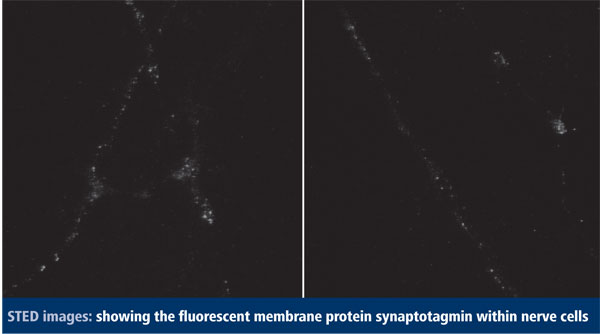
Rizzoli has used this technique to observe the movements of synaptic vesicles – those membrane bubbles that serve to release chemical messengers known as neurotransmitters in nerve endings. In 2008, Rizzoli, together with Hell and others, showed that the technique can record videos of vesicle movements at 28 frames/second with a spot size of 62nm.1
In a paper now in press,2 Rizzoli and coworkers study the movements of vesicles in living nerve cells in more detail, allowing them to follow the fates of many individual vesicles as they move around and become active or inactive, which was beyond the capability of all other methods available before. ‘Synaptic vesicles are small (~40nm in diameter) and heavily packed, in clusters of ~200-500 nm in diameter,’ Rizzoli explains. ‘To “see” vesicles moving with resolution limited to a blurry spot of at least 200 nm in diameter requires labeling only single vesicles [which is not easy], or the use of other techniques, such as fluorescence correlation spectroscopy – where the objects are not actually “seen”. Thus, the real movement of these vital organelles was never investigated until the advent of super-resolution.’
Apart from STED, there are also a few other methods now that manage to bypass Abbe’s diffraction barrier with different tricks, including STORM (STochastic Optical Resolution Microscopy), which is now commercially available from Nikon; the closely related PALM (PhotoActivated Light Microscopy); and 4Pi microscopy.3
Look, no label!

At the end of the 1990s, Xie’s group introduced CARS (Coherent Anti-Stokes Raman Scattering) microscopy.4 Unlike ordinary light scattering, which you can observe when a ray of light crosses a room with smoke or suspended dust particles, Raman scattering involves a wavelength shift in the scattered light, in other words, a transfer of energy between the incident radiation and the scattering particle. As only a very small fraction of any light scattered exchanges energy, Raman methods typically involve powerful lasers and often some chemical enhancement as well.
In CARS, Xie explains, ‘the sensitivity is much higher than in conventional Raman microscopy, because the coherent excitation of vibrational oscillators in the sample results in a strong signal.’ Still, one needs high concentrations of the chemical bonds whose vibrations are observed. Lipids with their numerous carbonhydrogen bonds proved suitable targets for this technique, which allowed the non-destructive, label-free imaging of lipid vesicles. Harvard has licensed the CARS technology to Carl Zeiss MicroImaging for implementation in its confocal microscopes.
In 2008, Xie, together with his graduate student Christian Freudiger and postdoc Wei Min presented a new, more efficient Raman-based imaging method, known as SRS (Stimulated Raman Scattering), which enabled them to trace medically relevant structures and movements in living cells with extremely high sensitivity. ‘SRS spectroscopy existed previously, but we developed it into a high sensitivity imaging method with a moderate laser power tolerable by biological samples, and overcame several major difficulties in CARS microscopy, which include nonlinear concentration dependence, complex image contrast interpretation, and distorted spectra that are hard to assign to chemical species,’ Xie notes. For instance, they could directly observe the distribution of omega-3 fatty acids in living cells, and monitor the uptake of drug molecules through the skin.5 The technology has been licensed to instrument makers and Xie hopes that commercial instruments applying his method will reach the market later this year.
In their most recent innovation to the field of label-free imaging, Xie, Min and graduate student Sijia Lu have now harnessed the very same stimulated emission effect that serves both to power lasers and to deplete excited fluorophores in STED. Unlike in STED, where the additional photon created during de-excitation is filtered out, Xie’s new technique, known as stimulated emission microscopy, picks up this enhanced radiation and uses it as evidence that the laser focus has hit one of the molecules it was looking for.6
This way, Xie and colleagues were able to detect even very low concentrations of a range of light absorbing molecules – chromophores – that did not display any detectable fluorescence. ‘Since these molecules do not fluoresce, they have literally been overlooked by modern optical microscopes,’ Xie says. In their first demonstrations of the new technique, Xie’s team showed that it can produce pictures of haemoglobin in living cells or tissues, of the distribution of certain proteins in live E. coli cells and the red blood cells in the vascular network of a mouse ear. With conventional methods, the imaging of capillaries would have required a contrast agent.
In a comment on Xie’s work, STED inventor Stefan Hell and his colleague Eva Rittweger suggest that the two different functions of stimulated emission, in other words, the depletion of excited molecules and detection of these very same molecules, could be combined in one super-powered technique. ‘An intriguing possibility for the future would be to design a set of laser pulses that fulfil both roles of stimulated emission […] to provide images of unlabelled, non-fluorescent molecules at sub-diffraction (nanoscale) resolution for the first time,’ they write.7
Xie is also optimistic that the technology can be developed further. ‘This is just the beginning,’ he comments. ‘Many interesting applications of the new imaging modalities are forthcoming.’
Complementary methods
With all these advances in optical microscopies, where does that leave electron microscopy (EM), traditionally the method of choice to image structures too small to be resolved in a light microscope?
While the conditions of EM, involving metal staining and high vacuum, rule out the direct investigation of living cells, this technology has also experienced improvements in the last years. For instance, the group of Hong Zhou at the University of California at Los Angeles, US, has recently elucidated the entry mechanism of a nonenvelope virus by solving its structure to atomic resolution using only cryo-electron microscopy.8 While some viruses, such as influenza or HIV, are wrapped in a membrane that can fuse with the cell membrane, the cell entry of those viruses lacking a membrane depends on structural changes in viral proteins, which were beyond the reach of previous investigations.
By flash-freezing the modified, entry-ready virus and pushing the structural analysis by cryo-EM to its technical limits, the researchers could detect the structural changes that enable the virus to enter the host cell. ‘This is the first study to determine an atomic resolution structure through cryo-EM alone,’ claimed post-doctoral researcher Xing Zhang, the first author of the paper, adding that ‘by proving the effectiveness of this microscopy technique, we opened the door to a wide variety of biological studies’.
Even though advanced light microscopies are moving into those parts of the length scale that hitherto only EM could reach, researchers feel that there is still plenty of room for both approaches to explore previously inaccessible reaches of the nanoworld.
‘[EM] is still the highest resolution technique for “seeing” objects in the context of the cell, and will undoubtedly remain the gold standard for investigating structures in fixed cells,’ Rizzoli comments. ‘All super-resolution techniques strive to investigate the behaviour of small structures in living cells, and therefore should be seen as complementary to electron microscopy, rather than as competitors.’
Michael Gross is a science writer based in Oxford, UK.
References
1. V. Westphal et al., Science, 2008, 320, 246.
2. D. Kamin et al., Biophys. J., 2010, in press.
3. T. Lang and S. O. Rizzoli, Physiology 2010, doi 10.1152/physiol.00044.2009
4. A. Zumbusch et al., Phys Rev. Lett., 1999, 82, 4142.
5. C. W. Freudiger et al., Science, 2008, 322, 1857.
6. W. Min et al., Nature, 2009, 461, 1105.
7. S. W. Hell, E. Rittweger, Nature, 2009, 461, 1069.
8. X. Zhang et al., Cell, 2010, 141, 472.



