
While TB is no longer a menace in the developed world, 9m new cases are reported each year with almost 2m deaths, particularly in countries where poverty and overcrowding are prevalent. It is a hard disease to eradicate since the causative organism – the bacillus Mycobacterium tuberculosis – is harboured by one third of the population: around 2bn people. This latent infection can be converted into active TB if the immune system is weakened by malnutrition, cancer or HIV/AIDS, and this co-occurrence of TB and HIV/AIDS is a rapidly growing problem as is the emergence of drug resistant strains of M. tuberculosis.
Current drug therapy
It is 60 years ago since Selman Waksman received the Nobel prize for physiology or medicine for his discovery of the first TB drug – the aminoglycoside antibiotic streptomycin, used from 1946 onwards. This discovery showed, for the first time, the value of systematic screening of microorganisms – Streptomyces in this case – for new antibacterials, an initiative that led to other aminoglycoside antibiotics, the tetracyclines erythromycin and vancomycin.
Streptomycin was initially used in combination with the synthetic drugs isoniazid, pyrazinamide and ethambutol, which had been discovered during anti-bacterial screening programmes, but was replaced in 1967 by the macrocyclic antibiotic rifampicin, another Streptomyces-derived drug. Both drugs interfere with mycobacterial protein synthesis, but in different ways. Streptomycin causes misreading and premature termination of translation of mRNA, while rifampicin binds to mycobacterial DNA-dependent RNA polymerase and inhibits the production of RNA.
The current therapy for TB, which has a cure rate of 85%, was launched in 1993 and involves using the four drugs – rifampicin, isoniazid, pyrazinamide and ethambutol – for the first two months, followed by rifampicin and isoniazid for the next four months. For patients who do not respond to this drug regime, one of the fluoroquinolone antibacterials such as ciprofloxacin, together with another of the aminoglycoside antibiotics such as capreomycin, kanamycin or amikacin, are prescribed.
The use of such a cocktail of drugs is tedious since it involves supervision, monitoring and an uninterrupted supply of at least four drugs for a six-month period, so patient compliance in developing countries is difficult to achieve. Moreover, there has been a worrying emergence of strains that are resistant to the two main drugs – rifampicin and isoniazid – particularly in the former Soviet Union, Southern Africa and China, where drug resistant TB affects up to 500,000 people annually. There have also been reports of ‘extensively drug-resistant strains’ of TB in at least 58 countries. These strains are resistant not only to rifampicin and isoniazid but also to pyrazinamide, ethambutol and various fluoroquinolones and aminoglycosides commonly used to treat TB. To this can be added the very recent appearance of a few cases of totally drug-resistant strains (TDR-TB) in Italy (2007), Iran (2009) and India (2012).
New drugs for treating TB are needed urgently. Ideally they should have a novel mode of action; be effective against both drug-sensitive and drug-resistant strains of M. tuberculosis and more rapidly eradicate the bacilli; be orally bioavailable; and have no unfavourable drug–drug interactions.
A tall order; however, the novel drug candidate TMC207 appears to satisfy most of these requirements.
TMC207
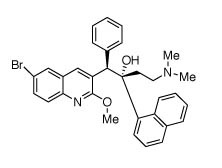
TMC207
Modern drug discovery comes down to designing novel molecular entities by matching structures to the known (X-ray) or presumed (computer modelled) structures of key enzyme targets. However, TMC207 and other novel diarylquinolines were not discovered using this strategy. They came from a screening programme that sought to identify compounds with activity against the bacillus Mycobacterium smegmatis, a distant relative of M. tuberculosis. There was no reason why compounds active against this organism, which is not pathogenic to humans, should have any effect against M. tuberculosis, but this leap of faith bore fruit.
In mouse studies, TMC207 inhibited both drug-sensitive and drug-resistant cultures of M. tuberculosis, but most significantly it also killed dormant, non-replicating, tubercle bacilli. In all these studies, TMC207 was as active as the drug combination of rifampicin, isoniazid and pyrazinamide, and when used with this drug triad also led to a more rapid clearance of bacilli.
In early clinical trials in patients with multidrug-resistant TB, there was a 48% cure rate after eight weeks when TMC207 was added to the drug cocktail of kanamycin, ofloxacin, ethionamide, pyrazinamide and cycloserine. In contrast, the cure rate for patients who received the five drugs without TMC207 was only 9%. In addition, there were no adverse side effects ascribed directly to this new compound.
This clinical study was particularly important since the patients all had advanced TB, which was resistant towards all of the standard drugs. More extensive and expensive clinical trials with less seriously affected patients are currently under way.
TMC207 works by inhibiting adenosine triphosphate (ATP) synthesis, specifically blocking the enzyme ATP synthase, and thus seriously curtailing the energy available to the organism. So a novel mode of action can be added to proven efficacy as reasons to be excited about this first new TB drug since rifampicin in 1967. Its relatively simple molecular structure should also allow optimisation of activity through the synthesis of analogues.
PA-824
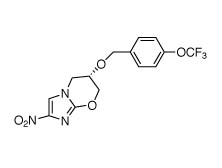
PA-824
Another experimental drug, PA-824, with a more intriguing mode of action is also in the pipeline. Under anaerobic conditions that mimic the dormant state of M. tuberculosis, this bicyclic nitroimidazole releases nitric oxide (NO), which inhibits a variety of enzymes involved in respiration. The drug acts as a respiratory poison similar to cyanide. Under aerobic conditions, as in active TB, PA-824 disrupts the growth of the bacterial cell wall.
The cell wall of M. tuberculosis is a highly complex structure, comprising an inner layer of peptidoglycan, similar to that of gram positive bacteria, and an outer layer made up of mycolic acids and arabinogalactan polysaccharides. PA-824 appears to inhibit the production of mycolic acids (cyclopropane-containing fatty acids). Since this type of lipid is not found in humans, this drug should have selectivity for M. tuberculosis.
The Global Alliance for TB Drug Development is currently running a Phase 2 clinical trial involving PA-824, pyrazinamide and the fluoroquinolone, moxifloxacin. In addition, more recent mouse studies have revealed that TMC207 used in combination with pyrazinamide and moxifloxacin also eradicates the bacilli.
These two triple drug regimes are particularly attractive since they do not include rifampicin or isoniazide – the two drugs to which the most resistant strains of M. tuberculosis do not respond.
Other biological targets
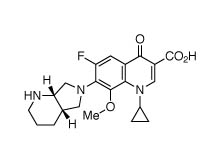
Moxifloxacin
Two other biological targets are the focus of preclinical studies for potential TB drug candidates. Mycobacterial DNA is usually found in a supercoiled state and the enzyme DNA gyrase controls the partial uncoiling of the DNA prior to replication and transcription. This class of bacterial enzymes has been successfully targeted for several decades by the fluoroquinolone antibacterials, and several new analogues, for example, moxifloxacin and gatifloxacin, are currently being evaluated for anti-TB activity.
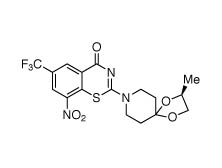
BTZ043
Growth of the arabinogalactan polysaccharide of the cell wall is the target of another promising drug candidate – the experimental benzothiazinone BTZ043. This compound inhibits the enzyme decaprenylphosphoryl-β-D-ribose-2’-epimerase that is needed for the formation of decaprenylphosphoryl-arabinose, which supplies the arabinose component of the arabinogalactan. Since arabinose is not a sugar used by human cells, there should again be selective destruction of mycobacterial cells.
In experiments with cells isolated from TB patients, BTZ043 displayed similar activity to the drug combination of isoniazide and ethambutol.
Genetic promise
Finally, in an effort to emulate the success achieved in cancer therapy by using drugs targeted at gene loci that are activated in cancer, many research groups are trying to identify loci that are associated with gene expression in active TB. For example, Luis Barreiro and colleagues recently reported (Proc. US Nat. Acad. Sci., 2012, 109, (4), 1204) that they had identified the gene locus DUSP14, which codes for the production of MAPK phosphatase.
The MAPK phosphatases are a large family of enzymes that dephosphorylate tyrosine and threonine phosphates on mitogen activated protein (MAP) kinases, rendering them inactive. This is of particular significance since the MAP kinases are linked to production of the cytokines TNF-α and interferon-γ, both of which are involved in immune protection against TB.
Clearly drugs designed to inactivate this locus or the phosphatases could enhance the immune system’s potential to destroy the bacilli. In addition, as Barreiro notes: ‘There is an inter-individual variation in susceptibility to TB, yet we still know little about the underlying genetic architecture.’ This work, for the first time, has revealed one gene locus that may increase susceptibility to development of full-blown TB.
So after decades of relative neglect, research on new anti-TB drugs is finally gathering momentum, not least due to the generosity and interest of agencies like the World Health Organization and the Bill and Melinda Gates Foundation. Funding from the latter is mainly directed towards the development of new vaccines, since the BCG is now only partially effective.
John Mann is emeritus professor of chemistry at Queen’s University Belfast, UK.



