
G. Richard Stephenson, University of East Anglia, UK
Probing the cycles of epigenetic control
In vivo chemical modification of nucleosides in DNA is part of nature’s control system to switch on and off the transcription of genes, in which the genetic code is read, and the corresponding protein is synthesised. Such chemical changes need also to be reversed so that a gene can be switched off again.
The two changes typically are not based on the same chemistry, so, for example, the methylation of deoxycytidine by DNA methyltransferase enzymes is reversed when no longer needed by oxidation and deformylation or decarboxylation. Thus, to reverse epigenetic methylation, as many as three steps are needed (1 to 2 to 3 to 4).
The presence of the hydroxydeoxycytidine (hmdC: 2) can lead to the repair of the DNA in which the hmdC is removed as an oxidised nucleoside, but the additional steps are advantageous since direct deformylation or decarboxylation returns the nucleoside to its original state without breaking the DNA strand, for which the repair can be unreliable and introduce errors.
To study the details of the direct processes, Carell’s group in Munich has introduced a fluorine label and the CH2 of the deoxyribose ring (Korytiaková, E. Kamińska, M. Müller, T. Carell; Angew. Chem. Int. Ed., 2021, 60, 16869).
With this probe, they were able to show that direct deformylation occurs in many different types of cells and monitor when it is occurring. For example, in neurons, almost all the deformylation has taken place during the first 48 hours of cell replication. Once replication has stopped, levels of 3 become stable.
The authors speculate that this residual 2-formyldeoxycytidine (3) may be important to promote condensation reactions between chromatin and histones.
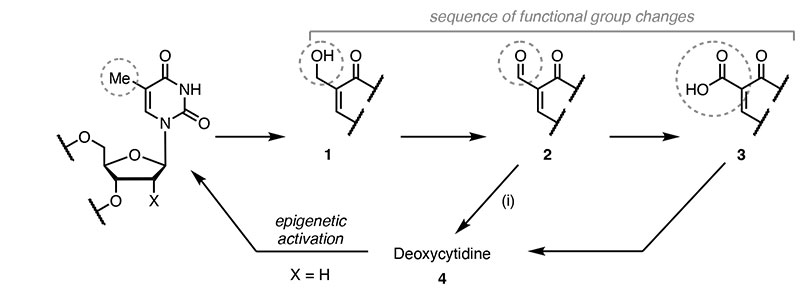
Scheme 1 The intermediates of a chemical genomic cycle for the nucleoside deoxycytidine (X =H) and synthetic fluorinated molecular probe (X = F); key intragenome functional group changed are ringed. Step (i) converts 2 directly into deoxycytidine to restart the epigenetic sequence by the action of a methyltransferase enzyme.
Electrophilic activation of amides
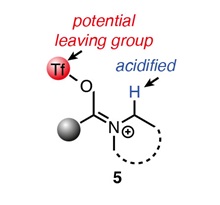
The key to this new approach (Figure 1) is to install a leaving group (currently triflate) on the oxygen of the amide, trapping an enol tautomer in which the N-alkyl positions of 5 become activated (P. Spieß, M. Berger, D. Kaiser, N. Maulide; J. Am. Chem. Soc., 2021, 143, 10524).
Figure 1 (right) An activated intermediate (deprotonation of 5 is promoted by transfer of electron density through the C=N double bond to detach a triflate leaving group (TfO‒).
Triflic anhydride (Tf2O) and the strong bulky base LiHMDS are employed together in this reaction. Some of the many examples reported in the paper are shown in Scheme 2.
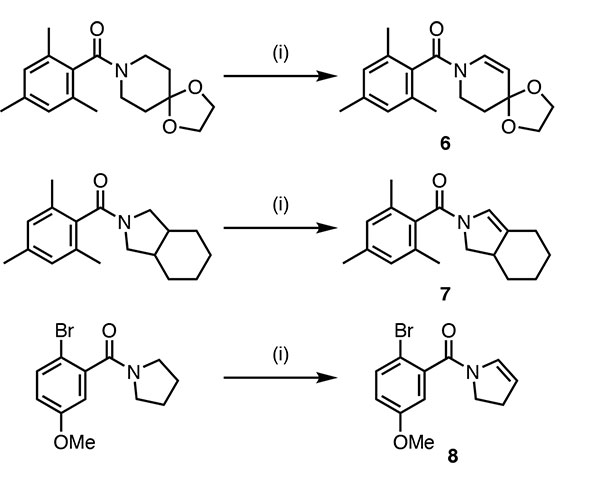
Scheme 2 Acylenamine synthesis [l:amide (0.30 mmol), LIHMDS (1.44 mmol, 1 M solution in THF, 4.8 equiv), then Tf2O (0.72 mmol, 2.4 equiv), ET2O (1.5mL)]
Differentiating enantiotopic diesters
Malonic esters are classic starting materials and can easily be made as derivatives with two different substituents at the central carbon atom by a sequence of two enolate alkylation reactions employing different alkylating agents. If the starting point was a symmetrical diester (ie RO2CCH2CO2R) dialkylation products such as 9 retain this symmetry and are prochiral.
Selective reaction at one of the two enantiotopic diesters will yield a non-racemic product, an approach that has, until now, been dominated by use of biotransformations employing esterases.
A non-enzymatic desymmetrisation method based on zinc-catalysed hydrosilylation has now been reported (P. Xu, Z. Huang; Nature Chem., 2021, 13, 634).
A range of chiral auxiliaries were examined and 11 (Scheme 3, box) was shown to give excellent results. The stoichiometry of the reaction was also investigated and a 2:1 ratio of Et2Zn and 11 gave the best yield and enantioselectivity.
The product ee rose dramatically from below 60% with equimolar amounts to nearly 100 % when increasing the excess of Et2Zn. Further increases in the excess of Et2Zn had little effect on ee but the reaction yield diminished.
In view of the unusual row of alkoxy, phenoxy and tert-amino functionality, a dizinc intermediate (10) was proposed as a key mediatory of the desymmetrisation process, so that the two zinc atoms help align the controlling stereogenic centre of the ligand and the prochiral features of the malonate diester substrate.
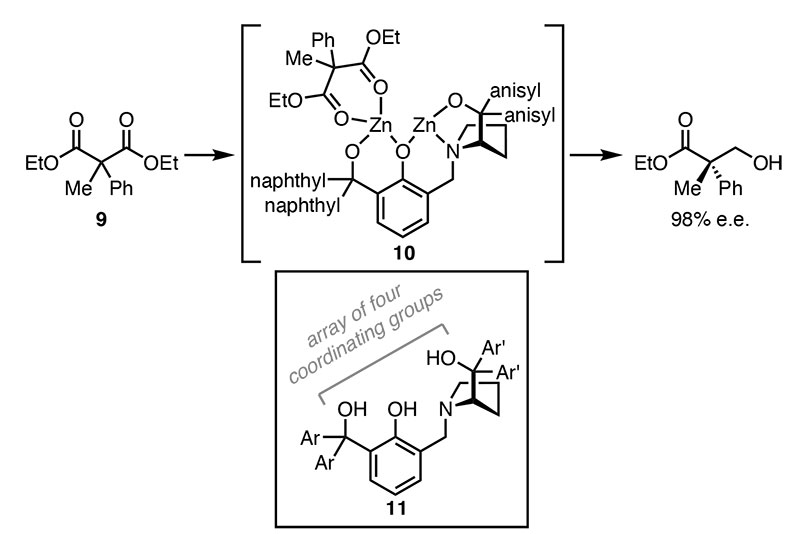
Scheme 3 Desymmetrisation using a chiral auxiliary bound to two zinc atoms (box: ligand 11 before addition of Et2Zn)



